|
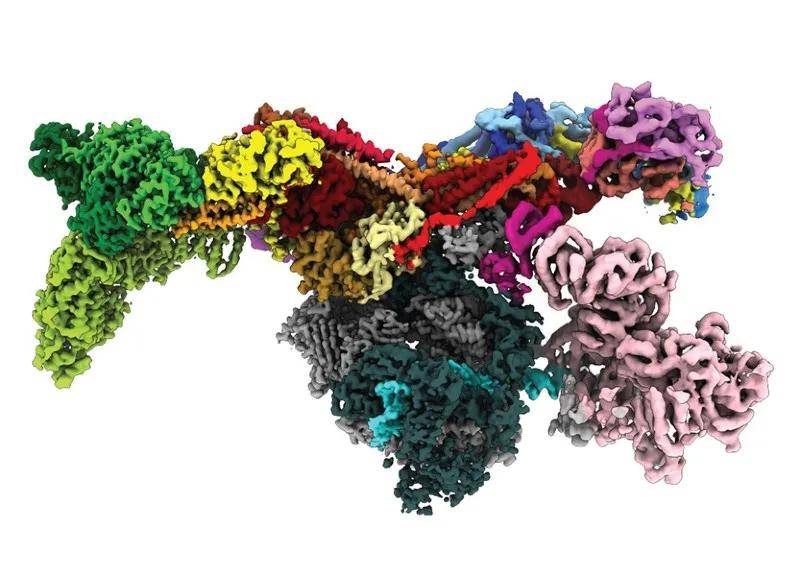
颠覆生命科学!AlphaFold预测完整人类蛋白质组结构神经网络AlphaFold的“颠覆性”数据库预测出了智人和20种模式生物的逾35万个结构。 人类基因组携带了逾2万个蛋白质的指令,但只有约1/3蛋白质的三维结构通过实验方法得到了解析,很多时候,这些蛋白质的结构只确定了其中一部分。
现在,一种名为AlphaFold的人工智能(AI)工具改变了现状。这款工具由位于伦敦的谷歌姐妹公司DeepMind开发,其预测的结构几乎覆盖了完整的人类蛋白质组(蛋白质组是一个生物表达的全部蛋白质)。除此之外,AlphaFold还预测了许多其他生物的几乎整个蛋白质组——从小鼠到玉米再到疟原虫(见“折叠选项”)。 这次预测的逾35万个蛋白质结构保存在一个公用数据库中,规模将在年底扩大到1.3亿个。虽然这些预测的准确度有高有低,但研究人员认为这些数据或为生命科学领域带来翻天覆地的变化。
“在我看来,这绝对是颠覆性的成果。”了解所有蛋白质的结构能让你弄清楚它们的机理。”伦敦大学学院(UCL)的计算生物学家Christine Orengo说。 “这是迄今为止AI在推动科学进步方面做出的最大贡献。我觉得这么说一点儿也不夸张。”DeepMind联合创始人、首席执行官Demis Hassabis说。 但研究人员强调说,这个数据泵只是一个开始,而不是结束。他们希望接下来能验证这些预测,更重要的是,将这些预测应用到目前尚无法实现的实验中。“拥有这个体量的数据是了不起的第一步。”伦敦大学学院计算生物学家David Jones说。Jones曾为AlphaFold的上一次迭代担任顾问。 获奖预测 去年,DeepMind在生命科学领域一鸣惊人——它的最新版AlphaFold在两年一度的蛋白质预测大赛CASP(蛋白质结构预测关键评估)中所向披靡。这个已举办多届的赛事向来是学术人员的竞技场,比赛要求参赛团队对已经通过实验解析但尚未公布的蛋白质结构进行预测。 AlphaFold的一些预测结果与准确度很高的实验模型相当,这让一些研究人员感叹AlphaFold将带来划时代的影响力。上上周,DeepMind发布了最新版AlphaFold的源代码,以及对其开发过程的详细阐述[1](许多学术团队已经开始利用这些资源进行重要预测)。在公开AlphaFold代码的准备工作中,DeepMind还对其做了优化,让代码运行起来更高效。CASP比赛中的一些结构曾让AlphaFold花了好几天的时间进行计算,但最新的AlphaFold只要几分钟到几小时就能完成计算。 效率的提升让DeepMind团队可以预测由人类基因组编码和20个模式生物的几乎所有已知蛋白。这些结构数据储存在英国EMBL-EBI(欧洲分子生物学实验室欧洲生物信息研究所)托管的一个数据库中。 AlphaFold预测的结构覆盖了98.5%的已知人类蛋白和其他生物的同比例蛋白,除此之外,AlphaFold还能评估其预测结果的可信度。DeepMind的工程师Kathryn Tunyasuvunakool说:“我们想让实验人员和生物学家清楚地知道,哪些预测部分是可信的。”Tunyasuvunakool是DeepMind发表在《自然》的描述蛋白质组预测论文的第一作者[2]。以人类蛋白质组为例,AlphaFold对58%的单个氨基酸位置的预测准确度足够高,可以用来判断蛋白质折叠形状,Tunyasuvunakool说。其中一部分预测——占整体的36%——的潜在准确度较高,或能揭示对药物设计有用的详细原子特征,比如酶的激活位点。 即使准确度稍低的预测结果也能带来重要信息。生物学家认为一大部分人类蛋白质和其他真核生物(细胞有细胞核的生物)的蛋白质拥有一些固有无序的区域,只是为了配合其他分子才形成了一些明确的结构。AlphaFold首席研究员John Jumper说:“许多蛋白质在溶液里扭来扭去,没有固定的结构。”AlphaFold预测的一些可信度不高的区域正好是生物学家认为无序的区域,DeepMind科学AI主管Pushmeet Kohli说。 研究人员认为,确定单个蛋白质如何与其他细胞组分相互作用是AlphaFold面临的最大难题之一。CASP大赛要求预测的大部分结构都是一个蛋白的独立折叠单元,也称为结构域。而人类蛋白质组和其他微生物蛋白质组的一些蛋白有多个半独立折叠的结构域。人类细胞还含有多个互作蛋白链组成的分子,比如细胞膜上的受体。 数据洪流 到今年年底,上周储存的约36.5万个预测结构将扩充至1.3亿个,这个数量接近人类已知蛋白总量的一半,EMBL-EBI的结构生物信息学家Sameer Velankar说。随着新蛋白的发现和预测能力的提高,这个数据库还会一直更新。Tunyasuvunakool说:“你本来不会相信有朝一日能获得这样的资源库。”她迫切想知道研究人员会带来哪些洞察。 研究人员已经在利用AlphaFold和相关工具理解来自X射线晶体学和冷冻电镜的实验数据。科罗拉多大学博尔德分校生物化学家Marcelo Sousa利用AlphaFold和细菌用来逃避抗生素黏菌素的蛋白质X射线数据来制作模型。实验模型中与AlphaFold预测结果不同的部分一般也是AlphaFold认为可信度不高的区域,Sousa认为这显示出AlphaFold能准确预测自己的极限。 不过,生物学家仍想将这些预测结果对照实验数据,从而对预测可靠性更有把握,英国MRC分子生物学实验室的结构生物学家Venki Ramakrishnan说。“我们需要能够信任这些数据。”Orengo解释道。 Jones对AlphaFold的能力印象深刻。但他认为AlphaFold预测的许多模型使用学界之前开发的软件也能计算出来。“之前软件给出的结果对于大部分蛋白质来说可能已经够好了,完全能够胜任一些研究工作。”那些下定决心一定要得到某些蛋白结构的研究人员利用实验方法或许也能成功。 不过,一下子多了这么多蛋白质结构,很可能会带来生物学的“范式转移”,美国哥伦比亚大学从事蛋白质结构预测的计算生物学家Mohammed AlQuraishi说。为了准确预测如此体量的蛋白质结构,他的领域付出了大量时间和精力,目前还没想好怎么利用这些资源。“之前用蛋白质序列做的研究,现在可以用蛋白质结构来做了。” Orengo希望这个数据库能帮她更好地理解蛋白质结构约束。她将一个已知蛋白质的数据库绘制成了5000个“结构家族”,但数据库中约一半的蛋白质只能被排除在外,因为这些蛋白无法在已经确定的结构中找到类似物。AlphaFold的预测结果或能揭示一些新结构,她说,“我们这下可以看到折叠空间的真面目了。” Jones预计AlphaFold会让许多生物学家思考要怎样利用这么多结构和可以轻松得到的更多结构。他说:“今后会有很多学术会议。我们现在有1.3亿个模型了,这会如何改变我们对生物学的认知?可能不会有改变,但我觉得会。” 参考文献: 1. Jumper, J. et al. Nature https://doi.org/10.1038/s41586-021-03819-2 (2021). 2. Tunyasuvunakool, K. et al. Nature https://doi.org/10.1038/s41586-021-03828-1 (2021). 原文以DeepMind’s AI predicts structures for a vast trove of proteins标题发表在2021年7月22日的《自然》的新闻版块上 |